蛋白质组研究的最新进展
电脑杂谈 发布时间:2020-05-30 23:10:11 来源:网络整理
蛋白质组学的概念最早是由马克·威尔金斯(Marc Wilkins)提出的,指的是基因组,细胞或组织表达的所有蛋白质. 蛋白质组学的概念和基因组的概念之间有许多差异. 它随组织甚至环境状态而变化. 在转录过程中,一个基因可以剪接成多种形式的mRNA. 蛋白质组不是基因组的直接产物. 蛋白质组中蛋白质的数量有时会超过基因组的数量. 蛋白质组学(Proteomics)处于早期的“发展”状态. 该领域的专家否认它是一种纯粹的方法,就像基因组学一样,不是封闭的,概念性的和稳定的知识体系,而是一个领域.
蛋白质组学专注于基因调控的动态描述,基因表达蛋白质水平的定量确定,疾病和药物对生命过程的影响的鉴定以及基因表达调控机制的解释. 作为一门科学,蛋白质组学研究,它不是从头开始的,它是蛋白质(多肽)光谱和基因产物作图技术的延伸,已有20多年的历史了. 肽图分析依赖于二维凝胶电泳(2-DE)和进一步的图像分析;基因产物图谱取决于各种分离分析,例如质谱分析,氨基酸组成分析等.
基于此,顾军向读者梳理了近年来蛋白质组学研究的最新进展.
1. 科学: 重大突破!人类蛋白质组亚细胞定位图的第一个定位
doi: 10.1126 / science.aal3321
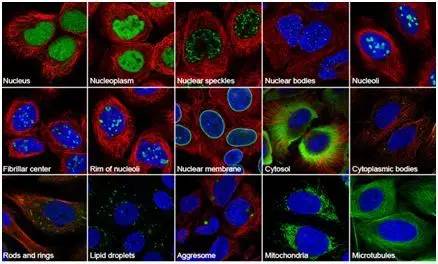
图片来自人类蛋白质图谱.
在一项新研究中,对蛋白质在人类细胞中的分布方式的首次分析显示,大多数人类蛋白质可以在给定细胞的多个位置找到. 研究人员使用瑞典的Cell Atlas,研究了人类蛋白质组的空间分布(与绝大多数蛋白质编码基因相对应),他们前所未有地描述了蛋白质在多个细胞器和细胞亚结构中的分布. 相关研究结果于2017年5月11日发表在《科学》杂志上,论文名为“人类蛋白质组学的亚细胞图”.
这项研究由瑞典皇家技术学院副教授艾玛·伦德伯格(Emma Lundberg)领导. Lundberg和他的团队生成了30万张图片,以系统地确定体外培养的细胞系中人类蛋白质的空间分布,并将它们定位在单个细胞分辨率下的细胞区域和亚结构中.
此细胞图是2016年12月启动的人类蛋白质图谱计划超过10年研究的结果. 这项新研究详细分析了这数十万张图片. 这些图片是国际合作行动的一部分. 这项国际合作计划还包括来自中国,韩国,印度,丹麦和德国的研究团队.
研究人员将总共13,003种抗体靶向的12,003种蛋白质靶向30个细胞区室和亚结构中的一个或多个. 此外,他们详细介绍了13个主要细胞器的蛋白质组. 蛋白质组最大的细胞器是细胞核(具有6,930个蛋白质)及其亚结构(如核小体和核斑)和细胞质(具有4,279个蛋白质).
2. 中国科学家建立了世界上第一个蛋白质组规模的健康人类尿蛋白定量参考范围,可用于健康监测和肿瘤筛查
doi: 10.1016 / j.ebiom.2017.03.028
在临床测试中,液体是血液以外最常用的体液样本. 从尿液中寻找新的生物标志物是临床蛋白质组学研究的热点之一. 然而,由于尿蛋白质组的巨大生理变异性和个体间差异,以及缺乏对健康人群中这些和差异的长期监测和系统评估,基于尿蛋白的生物标志物研究的假阳性结果非常高,基本上无法通过随后的临床验证. 5月2日,国家蛋白质科学中心(北京)的秦军研究小组在EBioMedicine上发表了题为“建立人类尿液蛋白质组监测生理和病理变化参考区间的概念验证工作流程”的研究论文.
本文的第一作者是副研究员冷文川. 该研究以两个国际中心的形式收集了来自167名健康志愿者的500个泌尿蛋白质组的数据. 系统地评估了差异,并在此基础上建立了世界上第一个健康人尿蛋白的蛋白质组规模的定量参考范围. 通过该定量参考范围分析跨大陆航班和肿瘤患者的尿蛋白质组数据,充分证明了尿蛋白定量参考范围在健康人群健康监测和肿瘤筛查中的重要应用价值.
3. 细胞: 重!历史上首次对完整的人类蛋白质组进行定量检测

doi: 10.1016 / j.cell.2016.06.041
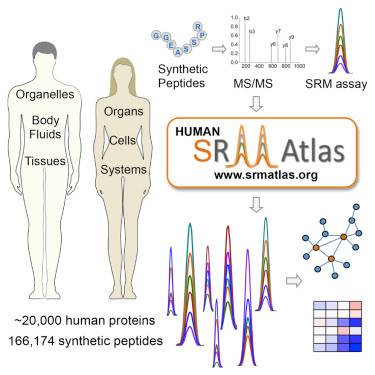
在一项新研究中,来自瑞士联邦理工学院(苏黎世联邦理工学院)和美国系统生物学研究所等机构的研究人员开发了人类SRMAtlas(人类SRMAtlas),这是针对性识别和高度重复定量预测的目录. 蛋白质组中所有蛋白质的特定质谱检测方法,包括许多剪接变体,非同义突变和翻译后修饰. 研究人员使用一种称为选择性反应监测(SRM)的技术,开发了使用166,174种易于理解的的蛋白质特征肽(蛋白质型肽)的检测方法. 相关研究结果发表在2016年7月28日的Cell中. 该论文的标题为“ Human SRMAtlas: 定量化完整人类蛋白质组的靶向分析资源”. 该论文的第一作者是美国系统生物学研究所的Ulrike Kusebauch博士. 通讯作者是美国系统生物学研究所的Robert Moritz教授和苏黎世瑞士联邦技术学院的Ruedi Aebersold.
SRMAtlas资源可从网站上免费获得,这将有助于公平地进行有针对性的,假设驱动的蛋白质组研究. 研究人员希望,该资源将大大加速基于蛋白质的实验室生物学的发展,以帮助理解疾病的转化和健康轨迹,因为从理论上讲,今天有可能鉴定和定量检测任何样品中的任何人类蛋白质.
可靠和可重复地检测任何组织或细胞类型的人类蛋白质组中任何蛋白质的能力触发了对系统水平的本质以及正常生理和疾病下特定途径的了解的变化. 在莫里茨教授的实验室中,研究小组能够使用SRM方法生成并验证由高度特定的目标蛋白质组检测方法组成的汇编目录,并通过这种广泛可用的灵敏且稳定的目标方法,质谱方法SRM可以定量检测99.7 20,277种标记的人类蛋白质中的%. 这种人类SRMAtlas可提供清晰的检测坐标,从而确定生物样品中蛋白质的特征肽.
4. 科学: 肝线粒体功能的系统蛋白质组学研究
doi: 10.1126 / science.aad0189
通过对小鼠进行蛋白质组学研究,科学家们对脂肪和能量代谢的分子遗传背景有了新的认识. 蛋白质组是生物体,细胞或组织的基因组中表达的所有蛋白质. 在一项新研究中,由瑞士联邦理工学院(苏黎世联邦理工学院)教授Ruedi Aebersold领导的团队,专门研究蛋白质组学,以及由洛桑瑞士联邦理工学院的教授Johan Auwerx领导的团队(EPFL) ),专门从事线粒体生理学和肝病研究的团队合作完成了这一突破性研究项目. 为了本研究的目的,蛋白质组是指在小鼠肝脏中表达的所有蛋白质. 相关研究成果已于2016年6月10日发表在《科学》杂志上. 该论文的标题为“肝线粒体功能的系统蛋白质组学”.
研究人员将来自大量老鼠的蛋白质数据综合在一起蛋白质组研究,以帮助他们解释其他代谢差异. 他们使用了称为SWATH-MS的质谱技术,该技术最近由苏黎世联邦理工学院的Aebersold团队开发. 它使研究人员能够测量该实验小鼠肝脏中一系列蛋白质的浓度.
研究人员研究了一组由40个小鼠品系组成的小鼠,这些品系可以追溯到两个相同的祖先,因此它们彼此密切相关. 来自这10个小鼠品系的代表性小鼠组形成同一组小鼠的多组,并向它们喂以高脂食品(即人们所描述的垃圾食品)或健康的低脂食品. 几周后,他们记录了这些小鼠的常规医学(生理)数据,尤其是测试它们的性能以及通过体育锻炼减轻体重的速度. 正如研究人员所预期的那样,这些小鼠对高脂食物摄入的反应不同. 有些老鼠患有脂肪肝等代谢性疾病,而另一些则没有.
为了进行评估,研究人员将这些生理数据与基因组,转录组和蛋白质组数据结合在一起. 从这些综合数据中,他们可以更准确地描述几种特定蛋白质在脂肪和能量代谢中的作用. 这些蛋白质之一是COX7A2L. 他们发现,在老鼠体内,这种蛋白质与线粒体中超复合物的形成有关,线粒体是细胞内的能量工厂. 这种超级蛋白质复合物由100多种不同的蛋白质组成,负责以合适的形式为细胞提供所需的能量. COX7A2L蛋白含量过低的小鼠无法提供足够的所需能量,从而对整个生物体产生负面影响.
5. 芝加哥大学化学评论学院赵颖明教授的研究组对组蛋白编码定量蛋白质组学进行了系统总结.
doi: 10.1021 / cr500491u

最近,芝加哥大学的赵英明教授的研究小组在国际顶级化学期刊《化学评论》(影响因子45.661)上发表了一篇题为“组蛋白修饰的蛋白质组学定量分析”的论文,对它进行了系统,深入的总结. 当前的组蛋白密码学定量蛋白质组学研究的前沿成果,并详细总结了20种400多种组蛋白代码. 其中,赵英明教授的研究小组率先发现了一半的组蛋白密码,这是目前世界上发现组蛋白密码最多的研究团队.
组蛋白代码包含基因序列和个体性状之间的关键调控信息. 它动态调节染色质的结构和功能,大大扩展了传统遗传密码的信息内容. 由于检测到蛋白质的定量变化,定量蛋白质组学越来越广泛地用于组蛋白代码的动态研究中. 目前,已经发现异常的组蛋白密码与许多主要疾病密切相关. 因此,发现了一种新的组蛋白密码,研究了组蛋白密码动态变化对基因功能调控的机理. 深入了解疾病的机理,靶向药物的开发甚至个体化治疗都具有重要意义.

本文系统地总结了利用生物质能谱和定量蛋白质组学解决组蛋白密码学领域问题的策略,技术和进展,并将该方法扩展到了全细胞非组蛋白等许多领域蛋白质修饰的鉴定,蛋白质修饰的调节酶底物的鉴定,蛋白质修饰的结合蛋白的鉴定等. 这项工作是由赵英明的研究团队和宾夕法尼亚大学的本杰明·加西亚教授的研究团队共同完成的. 黄河博士是本文的第一作者,而赵应明教授是通讯作者.
6. Anal Chem: 董梦球等人开发了定量蛋白质组学数据分析软件pQuant
doi: 10.1021 / a04246w
中国科学院pFind研究团队和我所董孟秋实验室开发了定量蛋白质组学数据分析软件pQuant,该软件使用计算方法消除干扰信号的影响,提高了肽和蛋白质定量的准确性,并量化每个值以进行准确性评估.
基于质谱的定量蛋白质组学是现代生物技术的增长点之一. 它用于测量在不同条件下复杂生物系统中蛋白质的丰度变化及其翻译后修饰. 研究蛋白质的功能和药物作用. 该机制的重要工具. 现有的定量软件通常无法有效消除干扰信号. 定量值的计算方法需要改进,并且缺乏准确性评估. 结果,输出结果是“鱼龙混”,由假阳和假阴引起的问题更加严重. 为了更好地解决这些问题,pQuant的开发者刘超(Liu Chao)研究了数百个可疑定量值的原始定量质谱和色谱数据,找到了原因和积累的经验,并充分利用了肽. 从肽定量到蛋白质定量的信号特征和方法,各种组合的灵活应用和统计算法,建立了非常详细的数据分析过程. 为了验证pQuant的性能,董孟秋实验室的宋春清用轻,重SILAC或14N / 15N标记哺乳动物细胞或细菌,以14: 1至1:10的不同比例混合14套标准样品,并生成14组测试数据. 组. 测试结果表明,在定量蛋白质组学领域蛋白质组研究,pQuant定量结果的准确性明显高于两个主流软件Census和MaxQuant,主要表现在: (1)pQuant输出的非数值比率的数量(即是,无法量化的部分)占总数的0.01-0.5%,远低于普查“ MaxQuant 2.5-10.7%和1.8-2.7%”的相应比率; (2)普查和MaxQuant输出许多不准确的结果,其定量值的标准偏差是1.3-pQuant的2倍; (3)pQuant给出了肽与蛋白质之间定量比的置信区间,而Census和MaxQuant没有准确度评估.
以上结果的标题为“ pQuant通过排除干扰信号并评估计算比率的准确性来提高定量”,并于2014年6月3日发表在《美国化学会》上的《分析化学杂志》上.
7. 自然: 科学家发布了人类蛋白质组草图的里程碑式结果
doi: 10.1038 / nature13319;土井: 10.1038 / nature13302
几天前,两个国际组织发表了《自然》杂志上人类蛋白质组的第一张草图. 这些在大多数非患者组织和器官中表达的蛋白质被用来更好地了解这种疾病. 人体在状态变化下奠定了坚实的基础. 这两项最新研究揭示了人类基因组的复杂性,并从以前认为属于非编码区的基因组中发现了新蛋白质.
本文来自电脑杂谈,转载请注明本文网址:
http://www.pc-fly.com/a/jisuanjixue/article-228151-1.html
-

-
油蔚
是赤裸裸的羞辱
加油